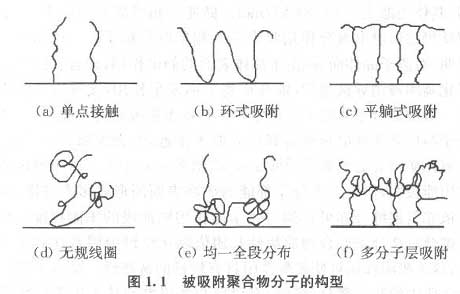
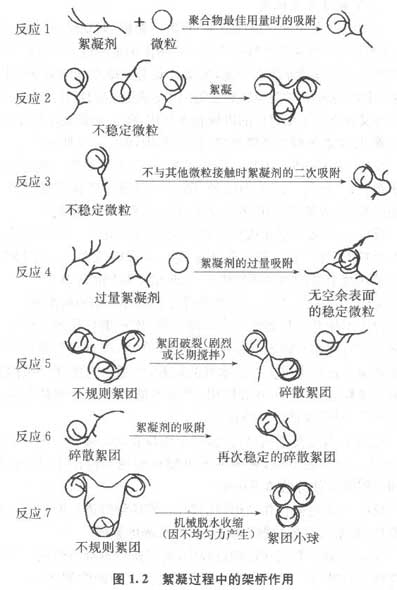
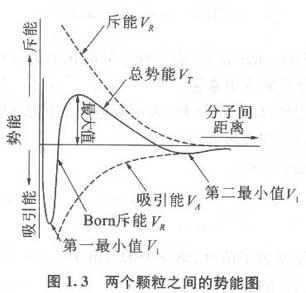
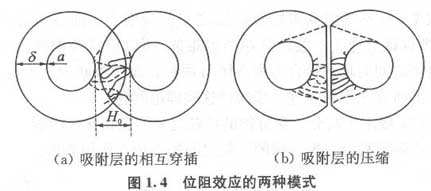
近30年来,国内外研究工作者在高分子絮凝剂研制、絮凝模式、絮凝工艺等方面进行了大量研究,并取得了一定的成果。
20世纪80年代,Ishibashi首次应用透射电镜研究水处理的混凝过程,把微观的絮体结构与形态观测同宏观的混凝现象结合起来进行综合分析,提供了研究混凝过程的一条新途径。且该方法根据胶体颗粒与所加混凝剂在水体中相互作用的真实结构来研究整个混凝过程,故能较准确地描述和解释混凝过程,因此引起有关学者的高度重视,并得到越来越广泛的应用。高宝玉等利用透射电镜技术研究了聚硅氯化铝(PASiC)的混凝机理,考察了搅拌时间和PASiC投量对混凝效果的影响,试验结果表明:随着搅拌时间的增长,形成的絮体逐渐增大;PASiC 用量的增加有利于形成大絮体而提高混凝效果。PASiC通过电性中和及吸附架桥作用使胶体颗粒脱稳。但仍然是一些定性结论而没有提取出混凝过程的微观结构。
综上所述,关于“架桥”絮凝机理的研究方法主要有两种。其一是基于实验室研究与理论计算,证实“架桥”的存在。其二是借助于高分辨率的透射电子显微镜镜直接拍摄“架桥”絮凝图像,但往往由于图像不清晰,没能直观地反映“架桥”模型。上述两种方法仅仅是定性地解释絮凝剂作用机理,不能深入絮凝过程的微领域,动态地研究絮凝过程,定量描述絮团结构。